SCoPE2
Shotgun single-cell proteomics method by Specht et al, 2019
Peer reviewed article: Specht, H., Emmott, E., Petelski, A.A. et al. Single-cell proteomic and transcriptomic analysis of macrophage heterogeneity using SCoPE2. Genome Biol 22, 50 (2021). 10.1186/s13059-021-02267-5
Data Websites
- Specht et al., 2019
- Petelski et al., 2021
- Leduc et al., 2021
- Montalvo et al., 2023
- Khan et al., 2023
- Khan, Elcheikhali et al, 2024
SCoPE2 is a second generation method improving upon SCoPE-MS and has a dedicated website at: scope2.slavovlab.net, which includes a detailed protocol.
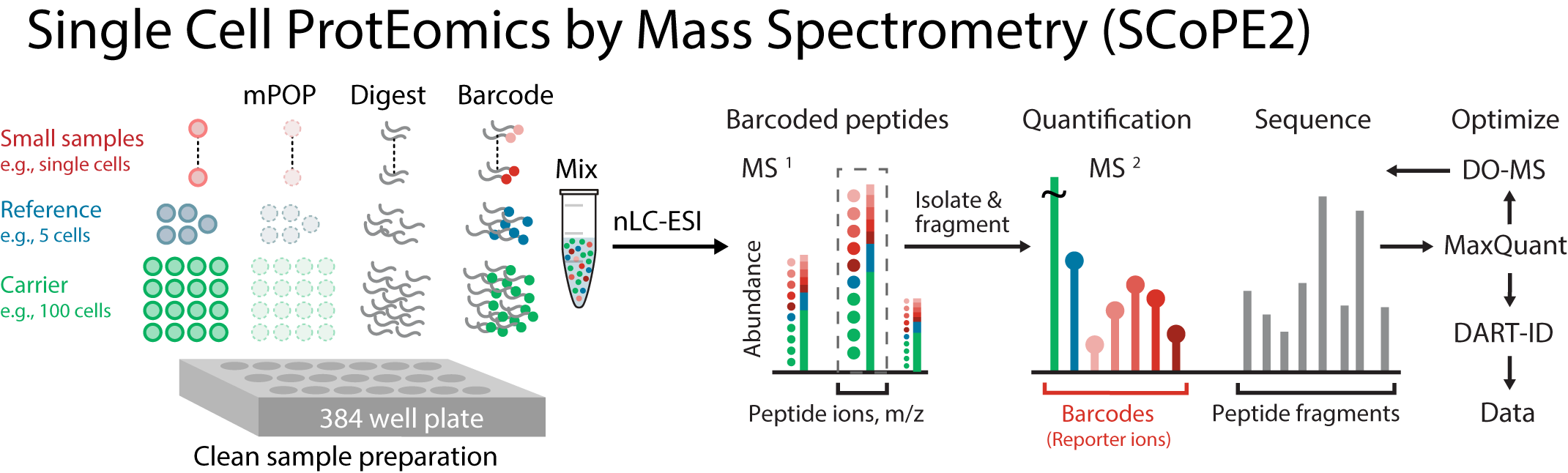
SCoPE2 introduced automated and miniaturized sample preparation that substantially lowers cost and hands-on time.
It uses data-driven analytics to optimize instrument parameters for sampling more ion copies per protein, thus supporting quantification with improved count statistics.
Furthermore, SCoPE2 uses peptide identification enhanced by incorporating retention time information within a principled framework, DART-ID.
SCoPE2 enables scalable, robust and affordable quantification of about 1,000 proteins per single cell, and about 3,000 proteins across many cells.
This coverage is achieved with 90 min of analysis time per SCoPE2 set (about 6 min / cell), which allowed us to analyze hundreds of cells on a single instrument in a couple of days.
Importantly, SCoPE2 succeeded in delivering and quantifying hundreds of ion copies from most detected proteins. This observation strongly supports the feasibility of single-cell LC-MS/MS protein quantification without amplification.
The fate and physiology of individual cells are controlled by proteins. Yet, our ability to quantitatively analyze proteins in single cells has remained limited. To overcome this barrier, we developed SCoPE2. It substantially increases quantitative accuracy and throughput while lowering cost and hands-on time by introducing automated and miniaturized sample preparation. These advances enabled us to analyze the emergence of cellular heterogeneity as homogeneous monocytes differentiated into macrophage-like cells in the absence of polarizing cytokines. SCoPE2 quantified over 3,042 proteins in 1,490 single monocytes and macrophages in ten days of instrument time, and the quantified proteins allowed us to discern single cells by cell type. Furthermore, the data uncovered a continuous gradient of proteome states for the macrophage-like cells, suggesting that macrophage heterogeneity may emerge even in the absence of polarizing cytokines. Parallel measurements of transcripts by 10x Genomics scRNA-seq suggest that our measurements sampled 20-fold more protein copies than RNA copies per gene, and thus SCoPE2 supports quantification with improved count statistics. Joint analysis of the data illustrates how variability across single cells can reveal transcriptional and post-transcriptional gene regulation. Our methodology lays the foundation for automated and quantitative single-cell analysis of proteins by mass-spectrometry.